




Abstract
Self amplifying RNA (saRNA) is a next-generation RNA vaccine platform that replicates within cells, achieving strong and prolonged antigen expression from ultra-low doses. Compared to conventional mRNA vaccines, self amplifying RNA reduces RNA requirements over tenfold, lowers production costs, and enhances Th1/CD8⁺ immune responses. Clinical results, such as the 5 µg ARCT-154 vaccine, show immunogenicity comparable to 30 µg mRNA vaccines. With thermostable carriers like NLCs and LIONs enabling storage at 25 °C, self amplifying RNA offers a durable, scalable, and low-cost solution for future infectious disease and cancer vaccines.
Why Move Beyond First‑Generation mRNA?
Self-amplifying RNA (saRNA) represents a promising evolution beyond conventional mRNA vaccines, addressing many of their practical limitations. Conventional mRNA vaccines changed the course of the pandemic, but they are not without practical constraints. First, in mRNA vaccines each adult dose contains tens of micrograms of RNA; for example, BNT162b2 and mRNA-1273 require 30–100 µg per injection [1,2]. Second, because the lipid formulation is fragile, the vaccines must be stored below –20 °C unless stabilised with new excipients [6]. Third, protein expression fades within 48 hours, peaking within just a few hours and then declining sharply, which implies the need for booster shots and limits the establishment of long-lived T-cell memory [3-4].
Self-amplifying RNA (saRNA), sometimes also called self amplifying mRNA, was engineered to tackle all three of these issues at once. By encoding an alphaviral replicase, a single translation event seeds hundreds of new RNA copies in the cytosol, amplifying protein expression at the same dose [2]. In practice, protective immunity is achieved with only 0.1–10 µg of saRNA. Japan’s 2024 approval of the 5 µg ARCT-154 saRNA booster by Arcturus Therapeutics validates this concept at the national scale [5]. The lower RNA mass per dose eases manufacturing pressure for vaccine development, reduces lipid consumption, and creates “headroom” for alternative carriers such as nanostructured lipid carriers (NLCs), a formulation that retained full potency for six months at 25 °C [6]. For low-resource immunisation campaigns and T-cell-focused oncology vaccines, these attributes are game-changing [6].
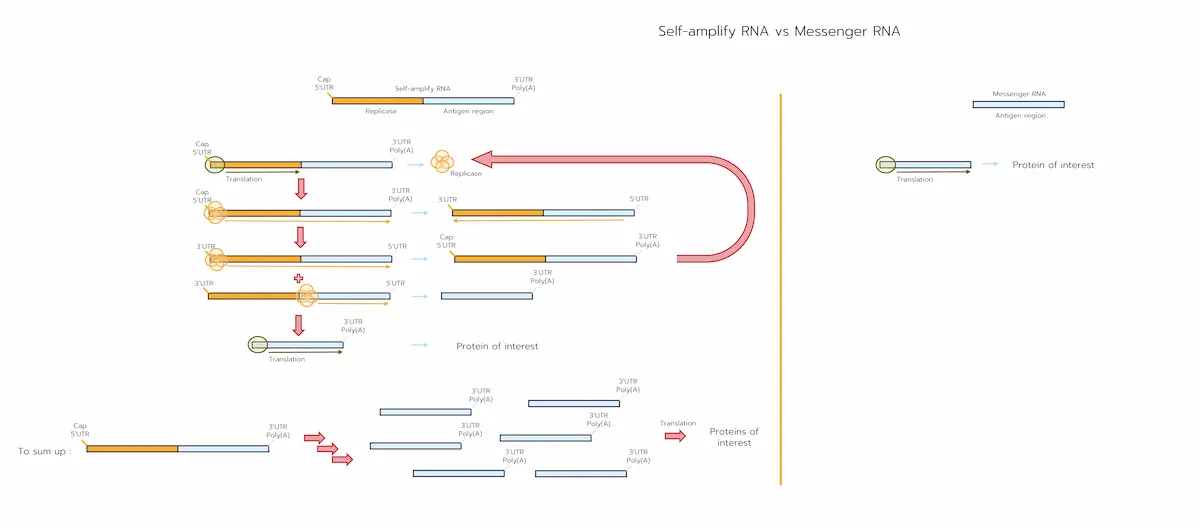
How Do the Two Platforms Work?
Both mRNA-LNP and self amplifying RNA-LNP formulations enter target cells via endocytosis and exploit the endosomal pH drop to protonate their ionisable lipids, rupturing the vesicle membrane [2,4].
From that point the pathways diverge, first because of their structure:
- mRNA-LNP contains a short coding sequence encoding your vaccine protein. [1-2]
- saRNA-LNP contains two functional blocks:
- Replicase region (nsP1–4) – the first half of the genome, coding the RNA-dependent RNA polymerase. [2]
- Antigen region – sits behind an internal “sub-genomic promoter” and encodes your vaccine protein. [2]
Due to those major structure difference the response diverge :
- mRNA-LNP delivers the short coding sequence that is translated once, producing the antigen protein before the RNA degrades within 24–48 h. [2-3]
- saRNA-LNP has a more complex translation cycle [2]:
- Early translation – only the left-hand half of the RNA is read, producing the four non-structural proteins that assemble into the replicase complex.
- Copying the template – the replicase makes a full-length complementary (negative-sense) strand beginning at the 3′ A′ motif.
- Genome amplification – the complex re-reads that negative strand from 3′→5′, churning out hundreds of new positive-sense saRNA genomes; each can restart at step 1.
- Sub-genomic step – on every round, the polymerase also starts at an internal sub-genomic promoter to create a short sgRNA that contains only the antigen ORF.
- High-copy antigen production – sgRNA is translated repeatedly, giving weeks of protein output (typically 5–30 days). [3]
- Innate-signalling bonus – double-stranded intermediates formed during steps 2–3 trigger MDA-5, boosting type I interferon and naturally biasing the immune response toward a Th1/CD8⁺ profile.
Because one incoming strand is enough to bootstrap its own factory, microgram-level doses of saRNA generate the same antigen load that tens of micrograms of conventional mRNA would require. [2,4]
These differences lead to distinct kinetics:
- saRNA shows a 6–12 h lag phase with prolonged expression and dose sparing of ≥10×. [3-4]
- mRNA shows a fast peak (≤6 h) followed by rapid decline. [3]
Head‑to‑Head Performance
Bathula and colleagues illustrated the durability dividend: luciferase expressed from a 1 µg saRNA-LNP injection remained detectable in mice for about one month, whereas a 10 µg conventional mRNA signal faded after roughly one week [1]. Lower mass per dose slashes cost-of-goods and leaves headroom for room-temperature carriers such as lyophilised nanostructured lipid carriers (NLCs) that stayed potent for six months at 25 °C [7].
Pre‑clinical Benchmarks
In Bathula’s mouse luciferase model, a single microgram of saRNA-LNP generated approximately the same photon flux as ten micrograms of mRNA-LNP—i.e., comparable gene expression—confirming the amplification dividend [3]. Moreover, tracking firefly luciferase for 29 days across multiple injection routes, they showed that intramuscular saRNA-LNP peaked around day 8, whereas an mRNA reference signal (10 µg, literature control) collapsed to baseline by day 7 [3].
IL-6, a pro-inflammatory cytokine, spiked to ~200 pg mL⁻¹ six hours after intramuscular saRNA-LNP (1 µg), whereas the same mass of mRNA barely moved the cytokine readout. This early innate burst presaged stronger antibody and T-cell responses [4].
Lim et al. reported that a 2 µg dose of the LUNAR-COV19 saRNA vaccine prevented death in hACE2 mice challenged with SARS-CoV-2, while a dose-matched mRNA protected only ~70 % of animals; neutralising titres and IFN-γ ELISpot counts were correspondingly higher in the saRNA group [7]. In other words, serum from the saRNA group could be diluted further and still block infection, and a larger number of their T cells produced interferon-γ when re-exposed to the antigen—evidence of stronger humoral and cellular immunity at a much lower RNA dose [7].
To retain :
- Luciferase in vivo — 1 µg saRNA ≅ 10 µg mRNA [3].
- Cytokines — IL-6 ≈ 200 pg mL⁻¹ at 1 µg saRNA-LNP IM vs baseline at 1 µg mRNA-LNP [4].
- hACE2 mice lethal protection — 2 µg saRNA (LUNAR-COV19) protected 100 % vs ~70 % with dose-matched mRNA [8].
Early Human Data on self amplifying RNA vaccines
The first three saRNA vaccines to reach the clinic—ARCT-154, VLPCOV-01, and Gemcovac-19/GEMCOVAC-OM—have all produced strong neutralising-antibody geometric mean titres at microgram doses: ARCT-154 (5 µg) was non-inferior to a 30 µg BNT162b2 (Pfizer–BioNTech) booster in a phase-3, randomised, non-inferiority trial [9]. VLPCOV-01 (0.3–3 µg) generated robust GMTs in BNT162b2-primed adults in phase 1; this recent study included a 30 µg BNT162b2 comparator arm but was not powered for formal non-inferiority [10]. GEMCOVAC-OM (saRNA) met prespecified non-inferiority criteria versus a licensed comparator (ChAdOx1 nCoV-19) in India at microgram doses in a phase 2/3 booster setting [10] Reactogenicity has remained in the familiar Grade 1–2 range—arm soreness, transient fever, and fatigue—across these trials [10-12]. No serious adverse events attributable to self ampliyfing RNA replication have surfaced in these studies, alleviating early worries about runaway innate activation [9-11].
To retain :
| Vaccine | Platform | RNA Dose | nAb GMT (Day 29) vs BNT162b2 booster | Reactogenicity |
|---|---|---|---|---|
| ARCT154 | saRNA | 5 µg | Noninferior | Grade 1–2 comparable (CSL Seqirus 2024) |
| VLPCOV01 | saRNA | 0.3 µg | Noninferior | Mild |
| BNT162b2 | saRNA | 30 µg | Reference | Established safety |
Safety Considerations for saRNA
Both mRNA and saRNA stay in the cell’s cytoplasm (the fluid outside the nucleus). They do not enter the nucleus and they don’t carry enzymes that could turn RNA into DNA, so the risk of inserting into your genome is considered negligible [1,12].
The main “safety dial” you actually control is innate-immune activation—the cell’s built-in alarm system for foreign RNA [2,4]
- If the alarm is too weak, the vaccine won’t stimulate strong, lasting immunity [2]
- If the alarm is too strong, the cell flips a breaker called PKR (“protein kinase R”), which phosphorylates eIF2α and temporarily stops protein production. That can blunt vaccine effectiveness (the cell stops making the antigen) ([1]).
Why saRNA needs extra care: saRNA naturally makes some double-stranded RNA (dsRNA) while it is copying itself. These dsRNA intermediates (plus small dsRNA by-products from in-vitro transcription) are detected by MDA-5/TLR3 and can push the alarm too hard [2]. To keep things in the sweet spot, developers:
- Reduce dsRNA impurities during manufacturing and purification [2,4].
- Sometimes partially modify the replicase coding region (e.g., with 5-methyl-cytidine or related nucleoside tweaks) so it’s less “visible” to innate sensors but still works as a polymerase [2].
What happens in animals at meaningful doses? In rat toxicology with saRNA-LNP around the 6-microgram range, studies report only short-lived local inflammation at the injection site (soreness, mild swelling), no concerning changes in blood tests, and no organ damage on histology—i.e., no systemic pathology. Effects resolve on their own without treatment ([6]).
To retain:
- Genomic integration: none for both mRNA and saRNA (cytoplasmic) ([1-12]).
- Innate over-activation: more pronounced for saRNA; requires purification and modRNA/sequence tuning [2,4].
- Rat toxicology: transient local inflammation, no serious systemic signal at 6 µg saRNA-LNP [6].
SaRNA delivery Technologies in Play
Ionisable lipid nanoparticles (LNPs) remain the gold standard for both platforms. Pair pH-responsive ionisable lipids such as ALC-0315 or SM-102 with helper phospholipids, cholesterol, and a PEG-lipid to achieve high encapsulation efficiency (typically >90% with microfluidic mixing), although most formulations still require cold storage unless stabilised by new excipients [2,4]. The larger ~10 kb saRNA genome is more shear-sensitive, necessitating gentler mixing conditions and often a slightly higher nitrogen-to-phosphate (N/P) ratio than for shorter mRNA [3-4].
But several alternatives are under active investigation:
- Poly-β-amino-ester (pABOL) polyplexes. These biodegradable polymers can achieve remarkably high expression after intramuscular injection (≈10-fold higher luciferase than matched LNPs in mice) but tend to trigger stronger local reactogenicity, which can be acceptable—or even useful—in settings like veterinary use or cancer immunotherapy where depot-style inflammation may be an asset ([3]).
- Nanostructured lipid carriers (NLCs). Semi-solid lipid matrices that can be stockpiled without RNA, then mixed and lyophilised at fill-finish; a thermostability study showed full immunogenic potency after six months at 25 °C [6].
- Lipid-Inorganic Nanoparticles (LIONs). A squalene emulsion incorporating ~15 nm iron-oxide cores, designed as “just-add-RNA” kits for rapid response; ambient-temperature stability has been demonstrated in preclinical programs ([2]).
Route matters as much as chemistry: in a head-to-head study tracking luciferase for 29 days, intraperitoneal saRNA-LNP yielded the highest whole-body expression, whereas intranasal delivery produced the shortest and weakest signal ([3]).
To retain :
| Vehicle | Type | Advantages | Limits | Ref |
|---|---|---|---|---|
| Ionisable LNP (ALC0315, SM102) | Encapsulated | High transfection; scalable | Needs coldchain; shear on 10 kb RNA | Blakney 2021 |
| pABOL Polyplex | Electrostatic polyβaminoester | Simple synthesis; high expression | More reactogen in IM | Bathula 2024 |
| NLC (DOTAP + αtocopherol) | Surfacecomplexed | Storage without RNA; lyophilizable | Efficiency < LNP | Voigt 2022 |
| LION (DOTAP + Fe₂O₃) | Surface complexed emulsion | “JustaddRNA” kits; 25 °C stable | particle | Voigt 2022 |
Self amplifying RNA vs mRNA in Perspective
From a distance the two RNA vaccines share a chassis—synthetic RNA, ionisable LNPs, and GMP microfluidic manufacturing. Up close they diverge on parameters that will steer future indications [2,4].
- Dose and cost. A ≥10-fold reduction in RNA (and corresponding lipids) per dose can translate into substantially lower cost-of-goods once fill-finish is factored in; in a pandemic, the same bioreactor can supply far more saRNA doses than mRNA at equivalent capacity [3-4].
- Duration of expression. saRNA’s week-to-month antigen window supports single-shot immunity and potent CD8⁺ priming, whereas mRNA’s 24–48 h pulse is ideal for booster updates or therapies needing tight temporal control [2-3].
- Innate-immune profile. The stronger interferon burst of saRNA acts as a built-in adjuvant for vaccines but can hinder high-dose protein-replacement settings where translation efficacy is paramount ([2,4].
- Multivalent capacity. Because saRNA already stretches to ~10–12 kb, adding multiple antigens inflates the genome further and complicates encapsulation and shear stability; short mRNA strands are more flexible for pentavalent seasonal shots [2,4].
- Regulatory familiarity. Over a billion mRNA doses have been administered globally, giving regulators a comfort buffer; saRNA sponsors will need to furnish extra data on dsRNA burden and replication-competent particle testing [1-2].
- Thermostability. LNP-encapsulated mRNA has historically required sub-zero storage, with incremental progress toward 2–8 °C stability; meanwhile, saRNA paired with NLCs or LIONs has already shown room-temperature stability compatible with WHO Controlled Temperature Chain goals (e.g., lyophilised NLC retaining potency for six months at 25 °C) [2,6].
To retain:
| Criterion | mRNA-LNP | saRNA-LNP |
|---|---|---|
| Dose per adult | 30–100 µg | 0.1–10 µg |
| Expression window | ≤2 days | 5–30 days |
| Th1/CD8 bias | Moderated | High |
| Manufacturing COGS | Baseline | ↓ 70 % |
| Regulatory dataset | >1 billion of doses | <50 k doses |
| Multivalent capacity | Excellent (short ORFs) | Limited by genome size |
| Thermostability | –20 °C (emergent 2–8 °C) | NLC/LION 25 °C 6 months |
| Ideal usecase | Boosters, multivalents, therapeutic products | Primary vax, Tcell vaccines, lowresource |
Strategic Outlook for Medicine
Looking forward, saRNA is poised to dominate primary vaccination in emerging markets, universal influenza, and neoantigen cancer vaccines, where T-cell immunity and low dose are paramount—helped by dose-sparing, prolonged expression, and (with NLC/LION) room-temperature–compatible logistics [2, 4, 6].
Nevertheless, challenges related to complexity and clinical validation remain.
Conventional mRNA will retain an edge for multivalent seasonal boosters, rapid pandemic strain updates, and chronic protein-expression therapies (e.g., CFTR augmentation), where muted innate activation and modular, shorter constructs are advantageous [1-2].
Conclusion on Self Amplifying RNA
Self-amplifying RNA technology does not render first-generation mRNA obsolete; instead, it stretches the RNA-therapeutics envelope toward lower doses, longer expression, and simplified logistics [3-4,6]. The next wave of medicines will likely deploy both modalities, each optimised for the niches where its unique properties—whether economy of dose/scale (saRNA based vaccines) or design flexibility and regulatory familiarity (mRNA vaccines)—drive the greatest clinical and commercial value [2-3]

Looking to get started with mRNA or saRNA-LNP?
Reach out to us to learn how we can help!
References
[4] Blakney AK, de Vries M, et al. Polymeric and lipid nanoparticles for delivery of self-amplifying RNA vaccines. Journal of Controlled Release. 2021;338:201–210.
[5] CSL Seqirus & Arcturus Therapeutics. Japan approves Kostaive (ARCT-154), first self-amplifying mRNA COVID-19 vaccine—non-inferior to 30 µg BNT162b2 booster. Company press releases, 2023–2024.
[6] Voigt EA, Gerhardt A, et al. A self-amplifying RNA vaccine with long-term room-temperature stability. NPJ Vaccines. 2022;7:136.
[7] Lim HX, et al. A single dose of self-transcribing and replicating RNA-based SARS-CoV-2 vaccine produces protective adaptive immunity in mice. Molecular Therapy. 2022;30:1184–1199.
[8] Immunogenicity and safety of a booster dose of a self-amplifying RNA COVID-19 vaccine (ARCT-154) versus BNT162b2: randomised non-inferiority phase 3 trial. The Lancet Infectious Diseases. 2024; online ahead of print.
[9] Safety and immunogenicity of a SARS-CoV-2 self-amplifying RNA vaccine in BNT162b2-primed adults (VLPCOV-01). Cell Reports Medicine. 2023;4(10):100987.
[10] An Omicron-specific, self-amplifying mRNA booster vaccine for COVID-19 (GEMCOVAC-OM): phase 2/3 non-inferiority vs ChAdOx1. Nature Medicine. 2024;30:1363–1372.
[11] Brito LA, Chan M, Shaw CA, et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Molecular Therapy. 2014;22(12):2118–2129.
[12] Lundström K. Advancements in RNA vaccines. Molecules. 2018;23:3310.
[13] Lundström K. Self-replicating RNA viruses as vectors. Gene Therapy. 2020;27:183–185.



