
Contact
Any inquiries about your nanoparticle/RNA-LNP formulation?
Drop us a line!

Contact
Any inquiries about your nanoparticle/RNA-LNP formulation?
Drop us a line!
Book a demo
Tell us about your nanoparticle or RNA-LNP project and request a TAMARA demonstration.
Our team will get back to you shortly to schedule your session.
Introduction
Lipid nanoparticles (LNPs) have emerged as the leading delivery platform for nucleic acids, enabling the clinical translation of messenger RNA (mRNA), small interfering RNA (siRNA), and other oligonucleotide-based therapeutics. (1,2) Their potential was most clearly demonstrated by the rapid development of mRNA vaccines, which highlighted the scalability, safety, and therapeutic efficacy of LNP-based systems. Beyond vaccines, LNPs are now being actively explored for applications including gene editing, protein replacement therapies, and cancer immunotherapy.
LNPs have become the preferred platform for nucleic acid delivery thanks to their intrinsic ability to protect fragile nucleic acids from enzymatic degradation, improve cellular uptake, and enable efficient cytoplasmic release. (3) Their tunable physicochemical properties (e.g., particle size, surface charge), together with a modular lipid composition, allow precise control over pharmacokinetics and functional performance, while enabling customization for diverse therapeutic applications.
An LNP typically contains four key components (4,5):
- Ionizable lipids (40 – 50 mol%) – the most critical LNP component. With an acid dissociation constant (pKa) around 6.0–6.5, these lipids become positively charged at acidic pH, allowing them to efficiently encapsulate negatively charged RNA during LNP formation. Once inside cells, they trigger release of RNA into the cytoplasm by destabilizing endosomal membranes through interactions with negatively charged endosomal lipids. Being relatively neutral at physiological pH, these lipids reduce cytotoxicity. (6–8) Notable examples include DLin-MC3-DMA (Patisiran), SM-102 (Moderna), ALC-0315 (Pfizer-BioNTech), LP-01 (Novartis), and C12-200 (research).
- Helper phospholipids (10 – 15 mol%) – support membrane structure. Depending on the lipid used, these lipids can enhance intracellular RNA release or improve in vivo circulation time. (7,9) Common examples include DSPC (used in Patisiran, Moderna, Pfizer-BioNTech) and DOPE.
- Cholesterol (38 – 50 mol%) – enhances the stability of lipid membrane, reduces interactions with serum proteins, and modulates membrane fluidity. (7,9)
- PEG-lipids (1.5 – 2 mol%) – form a protective hydrophilic layer on LNPs, improving stability and circulation time, reducing aggregation, and helping control particle size and distribution. (7) Some examples include PEG-c-DMG (Patisiran), PEG-DMG (Moderna), and ALC-0159 (Pfizer-BioNTech).
Efficient manufacturing is essential to fully exploit the potential of LNPs. Microfluidic production enables highly reproducible synthesis with precise particle size control and excellent encapsulation efficiency. Inside Therapeutics’ TAMARA, a plug-and-play microfluidic platform, provides these features in a user-friendly format, supporting applications from early-stage screening to in vivo studies (Fig. 1).

Materials
Table 1. List of equipment, reagents, and consumables required for RNA–LNP formulation.
| Category | Materials |
|---|---|
| Equipment | ● TAMARA formulation system ● Magnetic stirrer (if dialysis) or centrifuge (if centrifugal filter units) for buffer exchange ● Dynamic Light Scattering (DLS) for colloidal analysis ● Fluorescence microplate reader for RNA quantification (e.g., Ribogreen assay) |
| Reagents | ● RNA stock solution ● Lipid stock solutions (ionizable lipid, helper phospholipid, cholesterol, PEG-lipid) ● Ultra-pure ethanol for dissolving lipids ● Aqueous buffer for dissolving RNA (e.g., 10 mM citrate buffer, pH 4.0) ● Dilution buffer (e.g., Phosphate-buffered saline (PBS), 1X, pH 7.4) |
| Consumables | ● Eppendorf tubes and falcon tubes (15 and 50 mL) ● Dialysis membrane (e.g., Pur-A-Lyzer Midi Kit, MWCO 3.5 kDa) if dialysis ● Centrifugal filters (e.g., Amicon Ultra Centrifugal Filters, MWCO 100 kDa) if ultrafiltration by centrifugation ● 0.22-μm filter (e.g., PVDF) for sterilization |
Protocol for RNA-LNP production using TAMARA
Step 1: Preparation of lipid mix (Organic phase)
- Prepare individual lipid stock solutions in ethanol (e.g., 10 mg/mL) and store as single-use aliquots at −20°C to prevent repeated freeze–thaw cycles.
- Equilibrate lipid stocks to room temperature (≥20 min) prior to use. Ensure they are totally dissolved by gently warming, vortexing, or sonicating if needed.
- Transfer the required volumes of each lipid component, according to the predetermined molar ratios (e.g., SM-102:DSPC:Cholesterol:DMG-PEG2000, 50:10:38.5:1.5), into a single tube to form the organic phase. Finally, add the appropriate volume of ultra-pure ethanol to the combined lipid components to achieve the desired final concentration and total volume. Mix thoroughly by gently vortexing or pipetting to ensure a homogeneous solution.
- Required lipid and nucleic acid amounts can be rapidly calculated using our RNA-LNP Formulation Calculator (https://insidetx.com/resources/lnp-formulation-design-calculator/).
- For convenience and rapid development, we provide standard LNP Starter Kits containing pre-validated lipid compositions for RNA-LNP formulation (https://insidetx.com/product/lnp-starter-kits/).
Example: In this protocol, we illustrate LNP formulation using a 1 mL total volume per run at a flow rate ratio (FRR) of 3. Each run would then consist of 250 µL lipid mixture combined with 750 µL aqueous RNA solution. For triplicate formulations, a total of 750 µL lipid mix is required (3 × 250 µL). To account for minor handling losses, it is recommended to prepare slightly more than these volumes. In Table 2, we provide an example for preparing 1 mL of lipid mix, which is sufficient for all three runs and ensures there is extra volume available if needed.
Table 2. Example lipid mixture for LNP formulation. Preparation of SM-102:DSPC:Cholesterol:DMG-PEG2000 (50:10:38.5:1.5 mol%). Quantities are given for preparing 1 mL of lipid mixture. The total lipid concentration is 5 mg/mL; 200 µL of lipid mixture should be combined with 800 µL of ultra-pure ethanol to reach the final 1 mL volume.
| Lipid | MW (g/mol) | Molar ratio (%) | Mass required (mg) | Volume required (µL) |
|---|---|---|---|---|
| SM-102 | 710.2 | 50.0 | 2.86 | 114 |
| DSPC | 790.0 | 10.0 | 0.64 | 26 |
| Cholesterol | 386.6 | 38.5 | 1.20 | 48 |
| DMG-PEG2000 | 2509.2 | 1.5 | 0.30 | 12 |
Step 2: Preparation of RNA solution (Aqueous phase)
- Maintain RNase-free conditions to preserve RNA integrity. Use RNase-decontamination sprays to clean all work surfaces and pipettes, and handle all materials using RNase-free tubes, falcons, and tips.
- Determine the required RNA amount based on the target N/P ratio (e.g., N/P = 6). The N/P ratio defines the proportion of positively charged amine groups in the ionizable lipids to negatively charged phosphate groups in the RNA, which in turn sets the total RNA mass or volume needed.
- Thaw RNA stock solution, preferably on ice to minimize degradation.
- Prepare the RNA solution in 10 mM citrate buffer (pH 4.0), adjusting the concentration and total volume according to the calculation.
Example: Using the same 1 mL-per-run illustration at FRR 3, the total volume of RNA solution required for triplicate formulations is 2250 µL (3 × 750 µL per run). To ensure sufficient volume, we recommend preparing approximately 2500 µL of RNA solution.
Step 3: Microfluidic mixing
3.1. System setup
- Turn on the TAMARA formulation system and allow it to initialize.
- Ensure that the microfluidic chip and reservoirs are clean and completely dry before starting.
3.2. Pressure test
- If it is the first run, or if deemed necessary, perform a pressure test according to the TAMARA user guide (see p.23).
3.3. Chip design, solvent, and environmental parameters
Use the top-left corner menu of the TAMARA interface to set the chip design, solvent, and temperature.
- Confirm that the correct chip design is selected (Herringbone, the most commonly used micromixer; baffle design can be used as an alternative to achieve larger nanoparticles).
- Verify that the organic solvent is set to ethanol (or any other solvent you plan to use). In TAMARA, ethanol, isopropyl alcohol, acetone, and methanol are pre-set and can be selected directly. If you use a different solvent, it can be defined by entering its viscosity parameters. Note that nonpolar solvents such as dichloromethane or chloroform are not compatible with the cyclic olefin copolymer (CoC) microfluidic chips.
- Ensure the laboratory temperature is accurately set in the system before starting each run.
3.4. Microfluidic parameters
- Click “New Run” to set the microfluidic parameters.
Example:- Flow Rate Ratio (FRR): 3
- Total Flow Rate (TFR): 1 mL/min or 5 mL/min
- Total Volume: 1 mL
(!) FRR and TFR are critical parameters for tuning LNP characteristics and should be selected based on experimental goals. Higher aqueous-to-organic FRR or faster TFR generally produce smaller nanoparticles.
3.5. Reservoir loading
- Pipette the solutions carefully into the respective reservoirs:
- Aqueous phase (RNA in buffer): 750 µL
- Organic phase (lipid mix in ethanol): 250 µL
(!) Pipette gently to the center at the bottom of the reservoir to avoid bubble formation. Always load the aqueous phase first, followed by the organic phase (see p.29 of the TAMARA user guide).
3.6. Chip installation and collection setup
- Place the silicone gasket and the chip into the designated slot.
- Align the chip correctly to ensure the selected design is used.
- Close the system lid securely.
- Place a collection tube in the designated holder.
3.7. Run the experiment
- Start the experiment and collect the LNP sample in the collection tube.
- Monitor the experiment through the TAMARA interface and confirm that all parameters are correctly applied (parameters are displayed in the upper section of the screen).
3.8. System cleaning
- Place a 50 mL falcon tube in the holder as a waste collector.
- Perform at least one full cleaning procedure, consisting of a purge and cleansing with the recommended cleaning solutions.
- Run the recommended cleaning protocol according to the type of experiment:
- Between consecutive runs using the same lipid mix and RNA solution: Perform at least one washing sequence with ultrapure water in the aqueous reservoir and pure ethanol in the organic reservoir.
- Between runs with different lipid mixes or RNA solutions: Conduct a broader cleaning cycle. After the purge, run sequential washes as follows:
- Ultrapure water (aqueous) + ethanol (organic)
- Ultrapure water (aqueous) + ultrapure water (organic)
- Ethanol (aqueous) + ethanol (organic)
(!) Cleaning procedure may be adapted as needed depending on experimental requirements.
- After final cleaning, verify that the system and the chip are dry and in good condition for future runs.
Step 4: Post-processing
4.1. Buffer exchange
The final steps of LNP preparation involve buffer exchange to remove residual ethanol and replace the acidic formulation buffer with a more physiological medium, such as phosphate-buffered saline (PBS, pH 7.4). For small-volume preparations, dialysis kits or centrifugal filter units are commonly employed.
- Dialysis is a cost-effective method to remove residual ethanol. Commercial kits, such as Pur-A-Lyzer 3500 Da Midi kit, facilitate this process. In this method, the dialysis membrane is first rehydrated with the desired buffer according to the manufacturer’s instructions. The sample (typically 50–800 µL for Midi kits) is then carefully loaded into the membrane, ensuring proper sealing. The membrane is placed in a beaker containing sufficient PBS, allowing gentle stirring at room temperature or 4 °C overnight. Bath changes can be performed if necessary to ensure complete solvent removal. While effective and inexpensive, dialysis is relatively time-consuming.
- Centrifugal ultrafiltration offers a faster alternative for buffer exchange. This method employs Amicon Ultra centrifugal filters (100 kDa MWCO), with repeated rounds of centrifugation (e.g., three rounds at 3600 g for 30 min at 4°C) to concentrate the LNPs, remove residual ethanol, and adjust the pH to 7.4 with PBS. Although centrifugal filtration accelerates the downstream process, it is more costly compared to dialysis.
4.2. Sterilization & Storage
- Sterilization of the LNP suspension via 0.22 μm filtration (e.g., PVDF filter) is strongly recommended to prevent microbial contamination.
- LNP formulations can be stored at 4°C in PBS ideally under an inert atmosphere (e.g., nitrogen or argon) for up to one week.
Step 5: Characterization
Characterization is essential to ensure reproducibility, functionality, and quality of LNPs. Key attributes include particle size, polydispersity, surface charge, morphology, encapsulation efficiency and yield. These properties directly influence stability, delivery efficacy, biodistribution, toxicity, and ultimately therapeutic efficacy. Various analytical techniques are available to assess these parameters, each providing complementary insights. Table 3 summarizes commonly employed assays and the attributes they characterize.
Table 3. Common analytical assays for LNP characterization.
| Technique | Attribute measured/calculated |
|---|---|
| Dynamic Light Scattering (DLS) | ● Particle size ● Polydispersity index (PDI) ● Zeta potential |
| Ribogreen/Picogreen assay, QuantiFluor RNA/DNA kits | ● Free vs encapsulated RNA/DNA ● Encapsulation efficiency (EE%) and yield (EY%)* |
| Cryo-TEM | ● LNP morphology |
| High-Performance Liquid Chromatography (HPLC), mass spectrometry (MS) | ● Lipid identity, integrity, and quantity |
| UV–Vis spectrophotometry (Nanodrop) | ● RNA/DNA concentration and purity |
| Capillary electrophoresis (Bioanalyzer) | ● RNA/DNA integrity |
| Cell-based reporter assays, qPCR, Western blot, flow cytometry | ● Transfection efficiency, RNA translation, and downstream protein expression |
(*) EE% = [Encapsulated RNA]/[Total RNA (Encapsulated+Free)] x 100 ; EY% = [Encapsulated RNA]/[Input RNA] x 100. While EE(%) reflects the formulation performance, EY(%) provides a measure of both formulation and process efficiency.
Example formulation data
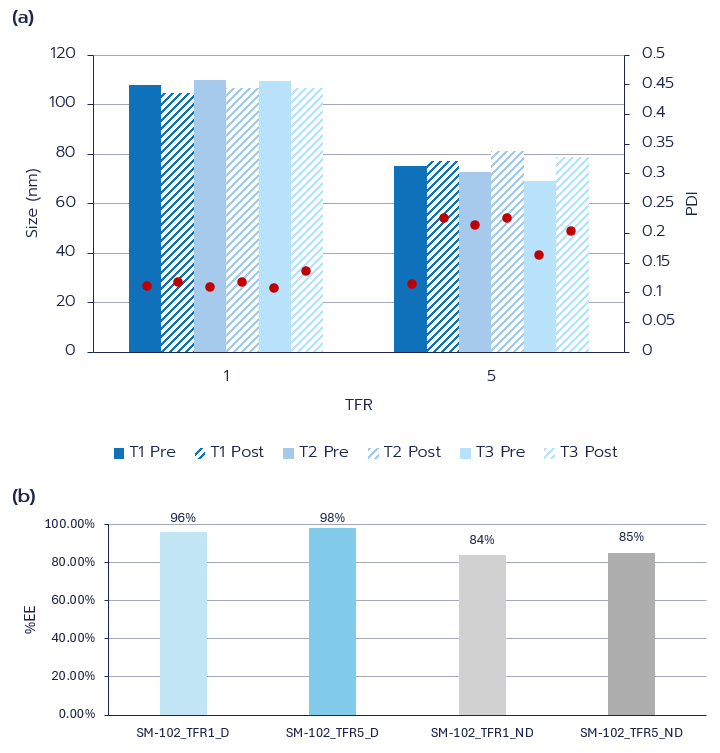
LNPs with a lipid composition like that of the clinically approved Moderna formulation (SM-102:DSPC:Cholesterol:DMG-PEG2000, 50:10:38.5:1.5) were synthesized on the TAMARA platform via microfluidic mixing (SHM) to encapsulate eGFP mRNA at an N/P ratio of 6. Formulations were prepared in 1 mL final volumes under varying flow conditions, with TFR set to 1 mL/min or 5 mL/min at a fixed FRR of 3, subsequently purified by dialysis, and characterized by DLS and Ribogreen assay.
At a TFR of 1 mL/min, the LNPs displayed uniform sizes ranging from 105–110 nm with PDIs below 0.20. Increasing the TFR to 5 mL/min produced smaller particles, averaging 69–81 nm, while maintaining a narrow size distribution (PDI≤0.20) (Fig. 2a). Encapsulation efficiency was consistently high, exceeding 80% across all formulations both before and after dialysis (Fig. 2b).
Acknowledgments
We wholeheartedly thank Nabila Laroui and Chantal Pichon from ART-ARNm (Inserm US-55) whose initial template served as groundwork for this protocol.
References
1. Hu B, Zhong L, Weng Y, Peng L, Huang Y, Zhao Y, et al. Therapeutic siRNA: state of the art. Vol. 5, Signal Transduction and Targeted Therapy. Springer Nature; 2020.
2. Fang E, Liu X, Li M, Zhang Z, Song L, Zhu B, et al. Advances in COVID-19 mRNA vaccine development. Vol. 7, Signal Transduction and Targeted Therapy. Springer Nature; 2022.
3. El Moukhtari SH, Garbayo E, Amundarain A, Pascual-Gil S, Carrasco-León A, Prosper F, et al. Lipid nanoparticles for siRNA delivery in cancer treatment. Journal of Controlled Release. 2023 Sep 1;361:130–46.
4. Kulkarni JA, Witzigmann D, Thomson SB, Chen S, Leavitt BR, Cullis PR, et al. The current landscape of nucleic acid therapeutics. Vol. 16, Nature Nanotechnology. Nature Research; 2021. p. 630–43.
5. Tieu T, Wei Y, Cifuentes-Rius A, Voelcker NH. Overcoming Barriers: Clinical Translation of siRNA Nanomedicines. Vol. 4, Advanced Therapeutics. John Wiley and Sons Inc; 2021.
6. Han X, Zhang H, Butowska K, Swingle KL, Alameh MG, Weissman D, et al. An ionizable lipid toolbox for RNA delivery. Vol. 12, Nature Communications. Nature Research; 2021.
7. Hald Albertsen C, Kulkarni JA, Witzigmann D, Lind M, Petersson K, Simonsen JB. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Vol. 188, Advanced Drug Delivery Reviews. Elsevier B.V.; 2022.
8. Kulkarni JA, Cullis PR, Van Der Meel R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018 Jun 1;28(3):146–57.
9. Cheng X, Lee RJ. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Vol. 99, Advanced Drug Delivery Reviews. Elsevier B.V.; 2016. p. 129–37.


Looking to get started or improve your LNP formulation screening?
Reach out to us to discover how we can help!