




Abstract
The lipid nanoparticle formulation system you choose does more than mix two liquids. It shapes the size, structure, and homogeneity of every particle you make, and those properties carry through to how your RNA behaves in cells and in animals. For researchers new to the field, the decision can feel like a single line item on a quote and that all systems look alike. They don’t.
This review walks through why the formulation method is a critical quality determinant, compares the two dominant technologies (impingement jet mixing and microfluidics), and lays out the practical parameters worth weighing before you commit, from volume range and reproducibility to the difference between encapsulation efficiency and yield. The goal is not to crown one system, but to help you match a platform to where your project actually stands.
Why the choice of lipid nanoparticle formulation system matters?
A lipid nanoparticle is not a fixed object you order from a catalogue. It is the product of a process, and the formulation step sits at the heart of that process. When an ethanolic lipid phase meets an aqueous phase carrying RNA, the RNA-LNP particles assemble in tens of milliseconds through nanoprecipitation and self-assembly. Everything about how that mixing happens (how fast, how uniformly, under what flow regime) sets the size, the internal organization (payload distribution, morphology…) and the surface presentation of the particles that result.
This is why the formulation method belongs upstream of almost every quality attribute that follows. Mixing conditions feed directly into the critical quality attributes (CQAs) of your LNPs (size, polydispersity, encapsulation, morphology, RNA integrity), and those attributes in turn govern stability, biodistribution, toxicity, and therapeutic efficacy. Change the formulation system and you change the inputs to that whole chain.

A recent benchmark made this unusually concrete. A Purdue University team systematically compared eleven mixing techniques (manual pipetting, several microfluidic mixers, and a range of turbulent-flow systems) while holding lipid composition, RNA, ratios, and buffer identical across every run. The only variable was the mixer. The particles that came out differed widely in size, encapsulation, internal structure, and biological performance. Same recipe, eleven different outcomes.
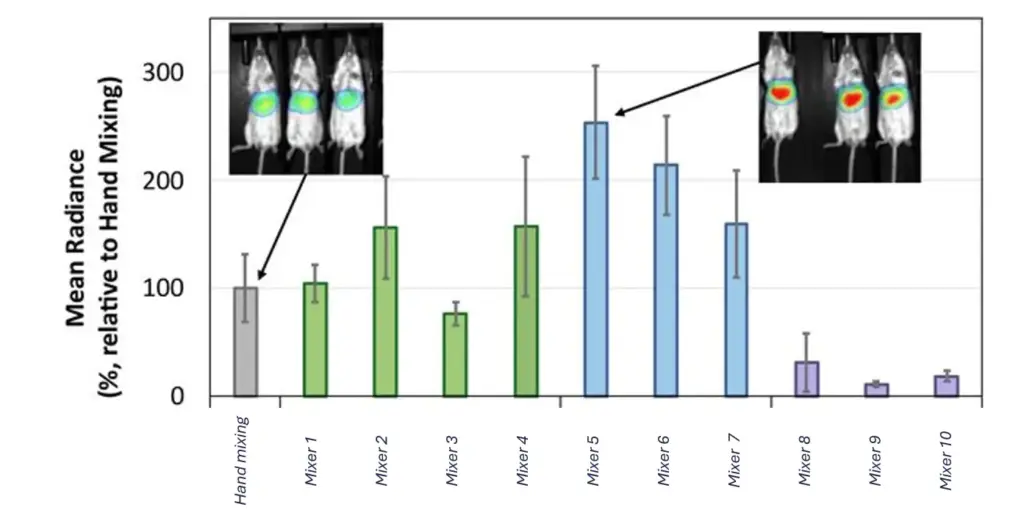
The practical lesson is blunt: the lipid nanoparticle is system-dependent. It is the product of a process, not a stable molecule you can order once and reproduce anywhere. An LNP is in fact metastable — the particles sit in a kinetically trapped state and keep evolving (fusing, maturing, aggregating) through downstream steps like dialysis and concentration. That is why the mixing conditions leave such a lasting mark on the final particle. It is a consequence that catches many newcomers off guard, and it deserves its own emphasis.
Your nanoparticles travel with your lipid nanoparticle formulation system
Because the particle is tied so tightly to how it was made, switching formulation systems means re-making your nanoparticles, not transferring them: even with identical lipids and ratios, the LNP you optimized on one platform will almost never reappear exactly on another. For early discovery this is an inconvenience; it becomes a real problem the moment your work has to move, from screening to in vivo, or later toward larger-scale production. And worth stating plainly: no single mixing technology today keeps the same nanoparticle across every scale of development. We come back to what that means for your choice at the end.
The starting point: hand mixing and its ceiling
Everything above is the scientific half of the decision. But choosing a system is also a practical decision (what volumes you can reach, how much RNA you lose, how easily you can run it), and that half is the one newcomers underweight. We weigh both below.
Most people meet LNPs through hand mixing: cheap, no equipment beyond a pipette, fine at very low volumes. Its ceiling shows up fast, though. Manual pipetting gives poorly defined mixing kinetics and depends heavily on who holds the pipette, producing larger and more variable particles and reproducibility that is hard to defend in a publication. In the Purdue benchmark, hand-mixed particles simply did not resemble those made by controlled systems, a comparison we unpack in our review on why handmade LNP can be limiting.
For anything beyond a first proof of concept at the corner of a table, the question becomes which controlled system to adopt. Before getting into the technologies, it helps to hold two kinds of criteria in mind at once, because a good choice has to satisfy both.
The first is scientific: which mixing technology, and therefore which particles, suits your application. This is where size, reproducibility, and the system-dependence discussed above live.
The second is practical: the volume range you can actually access, the losses you can tolerate, the cost per run, and whether the system is simple enough to run day to day. A platform can be scientifically excellent and still be the wrong choice if it wastes your RNA, locks you out of the volumes you need, or sits unused because it is too cumbersome. The sections below cover the technology question first, then the practical parameters, so you can weigh a system on both.
One thing ties the two together. Formulating LNPs is neither pure rational design nor blind screening, but a mix of the two. You reason about lipid composition, ratios, and process parameters to narrow the space, then you test empirically to converge, because the system-dependence of the particles means you cannot fully predict the outcome on paper. That testing is rarely hundreds of formulations, but realistically a handful to a few dozen conditions. And the moment you are running even that many, the practical criteria stop being secondary: a system that loses RNA on every run, charges you for a single-use cartridge each time, or takes an afternoon to operate makes iterative screening painful or unaffordable. Low losses, simplicity, and reusable consumables are what make the empirical half of the work actually doable.
A practical parameter checklist
Beyond the science of mixing, a handful of parameters separate a system that fits your work from one that fights it. The first is the mixing technology itself; the rest are the practical criteria that decide whether a platform is usable day to day. Here is what to actually look at.
1. Mixing technology: impingement jet mixing or microfluidics
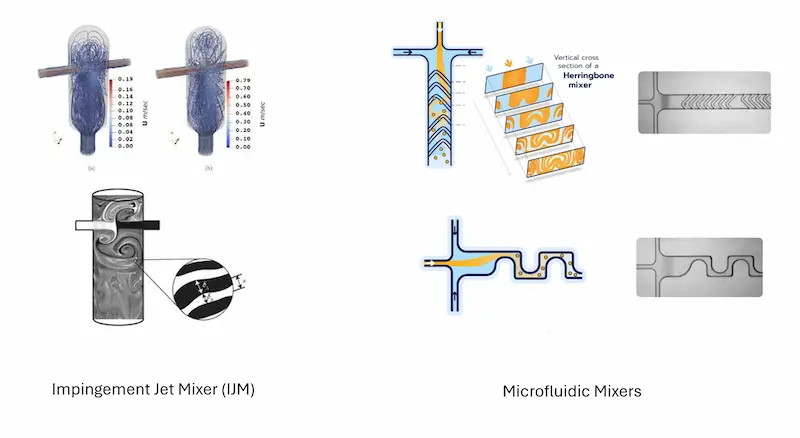
Controlled LNP formulation today is dominated by two approaches that solve different problems. Confusing them is the most common mistake newcomers make when reading a spec sheet, so it is worth knowing what each one demands.
Impingement jet mixing (IJM) (T-mixers, confined or multi-inlet variants) collides two high-velocity streams under turbulent flow. That turbulence is what makes the mixing near-instant and cleanly usable for continuous production, but it only works when the flow is fast (typically around 10 mL/min and up) and extremely well stabilized, hence not suitable for low volume production as it induces large losses and is difficult to scale up due to flow sensitivity. On top, flow rate ratio is generally locked at 1:1, otherwise mixing will occur outside the chamber, leading to degraded mixing performance and the particles suffer.
All these combined make IJM impractical for R&D, but perfectly suited for large scale production.
Microfluidic mixing combines the phases inside a defined channel geometry under laminar flow. Several mixing geometries can be used to influence mixing condition (herringbone mixer, baffle mixer…) hence giving flexibility on the characteristics of the produced nanoparticles. It gives fine control over particle formation through tunable parameters like flow rate ratio (FRR) and total flow rate (TFR), and runs on very small volumes, which matters when RNA is your most expensive reagent. Its constraint is the mirror image of IJM’s: laminar, diffusion-limited mixing does not scale up cleanly, with µm size channels prone to clogging and fouling at higher throughput while “scaling out” (running many channels in parallel) rarely is practical in reality.
| Impingement Jet Mixer (IJM) | Microfluidics | |
|---|---|---|
| Pros | Large scale production; very fast mixing; ideal at large volumes | Fine, tunable control; reproducible; Low RNA consumption; spans across preclinical stages |
| Cons | Impractical at small scale; large losses; heavy process; FRR fixed near 1:1. Scalability | Clogging/fouling cap scale-up; scale-out impractical. |
| Best for | Large-volume continuous production | Research and preclinical development |
Neither is “better.” They sit at opposite ends of the development timeline: IJM owns production, microfluidics owns research. For a newcomer working at research scale, microfluidics is almost always the right starting point.
2. Volume range
Match the system to the volumes you genuinely run, today and one step ahead. The amount you need scales sharply with your stage:
- Physicochemical characterization and in vitro work need very little. A few hundred microliters per condition (roughly 200–500 µL) is usually enough to cover DLS measurement (size & PDI), ribogreen assay (encapsulation readouts), and a cell assay.
- Mouse in vivo studies push you into the milliliter range. As a rough guide, you dose around 100 µL per mouse, several times per group, plus material for QC, so a few milliliters per formulation is a realistic working volume.
- Larger in vivo studies either on larger models (NHP) or larger cohorts can require tens of milliliters per formulation.
These are orders of magnitude, not fixed rules. The real figure depends heavily on your route of administration, the concentration you formulate at, and the indication you are working on, all of which change how much material a single study consumes.
| Development Stage | Indicative volume per formulation |
|---|---|
| Physicochemical + in vitro screening | ~200–500 µL per condition |
| Mouse in vivo (small cohort) | a few mL |
| Larger in vivo studies | tens of mL |
The practical trap is buying for where you are now: a screening-only platform leaves you stranded when you reach vivo and you’ll necessarily have to start from scratch all over again with it as the LNP your produce are different, while a production-scale system wastes majority of RNA at the bench. Ask what the realistic working range is, not just the headline number, and pick a system that covers the stage you are heading toward, not only the one you are in.
3. Encapsulation efficiency and encapsulation yield / formulation losses
Volume tells you how much material a study needs; losses tell you how much you actually have to make to get there. The two are inseparable: if a system wastes as much RNA as it delivers, the working volume on the spec sheet means little. That is where the two encapsulation metrics come in. They sound interchangeable. They are not, and you want to look at both.
Encapsulation efficiency (EE%) is the fraction of RNA that ends up inside particles, measured in your final sample (encapsulated RNA over total RNA present at the end). It matters because a high EE% means little free, unencapsulated RNA left in the formulation. Free RNA does not deliver, and residual free RNA can contribute to toxicity and unwanted reactogenicity, so you want EE% high (typically above 85–90%). Because most controlled systems clear that bar, EE% alone is a weak way to tell platforms apart.
Encapsulation yield (EY%) is the fraction of the RNA you started with that you actually recover encapsulated at the end. This is a process parameter: it reflects real losses (dead volumes, head-and-tail fractions discarded at the start and end of a run, material stuck in cartridges). A system can post a beautiful 95% EE% while quietly losing a third of your input RNA to waste, leaving a poor yield.
This is where many systems that are not truly optimized fall down: they lose a large share of the sample, and homemade platform and IJM in particular wastes material at small scale because of major dead volumes and the very high flow rates it needs. For anyone working with expensive or scarce cargo, EY% is often the number that decides whether a platform is affordable in practice. In general good Encapsulation Yield should be >85%
Always ask about both, and specifically about where the losses go.
You can learn more about EE% and EY% calculation in our encapsulation efficiency measurement protocol
4. Payload and lipid flexibility
One of the strengths of the RNA-LNP platform is how cargo-agnostic it is. The same formulation approach works across essentially the full range of nucleic acids: mRNA, saRNA, circRNA, siRNA, ASOs, and CRISPR components such as Cas9 mRNA with a guide RNA. Good news is, most platform are both lipid & RNA agnostic.
Note that if you are also interested in also making other types of nanoparticles (polymer-based, peptides…), it’s also important to consider solvent compatibility.
5. Reusable chips vs single-use cartridges
Both approaches have pros and cons. Single-use consumables are extremely useful to avoid cross-contamination, but over a year of running several formulations a week the cost compounds quickly, and you are locked into whatever the vendor charges per cartridge (and into their stock levels). Reusable chips, cleaned between runs, change that arithmetic substantially. This is less about science than about whether your formulation budget survives contact with reality, but for an academic lab on a grant it can decide whether the platform is sustainable at all. The full trade-off:
| Single-use cartridge | Reusable chips | |
|---|---|---|
| Cross-contamination | Strong — a fresh consumable every run | Relies on a reliable cleaning protocol between runs |
| Cost per run | High and recurring; vendor-set | Low after the initial chip |
| Supply risk | Dependent on vendor stock | None once you own the chip |
| Throughput | Fast — swap and go | Slower — cleaning adds time between runs |
| Reproducibility | Can suffer: single-use chips are often lower quality | Consistent when cleaning is well controlled |
| Long-term sustainability | Erodes the budget over time | Sustainable for routine, frequent use |
The honest read is that each side wins on one axis. What you ideally want is not one or the other but the best of both: a system that uses an affordable dedicated consumable (so you can isolate a run and avoid cross-contamination where the risk actually matters, like an in vivo study, by dedicating a clean chip to it) that is also reusable (so the cost per run stays low). A reusable chip you can clean, or set aside per project, gives you the contamination control of single-use without the recurring bill.
6. Reproducibility
This is the quiet dealbreaker. A system that makes lovely particles once but drifts batch to batch will cost you more in failed experiments than it ever saved at purchase. This is also exactly where homemade rigs and low-quality platforms lag behind: without tight control over flow and timing, their batch-to-batch variation balloons and the result depends heavily on the operator. Reproducibility is hardest of all at low volume: below about 1 mL, the smallest variations in handling, dead volume, and timing get amplified, and consistency falls apart precisely where newcomers do their screening. Look for reported batch-to-batch variation (a coefficient of variation under 5% is a reasonable bar) rather than a single hero result.
7. Ease of use — and the trap of over-automation
A system you cannot operate is a system you do not use. For a newcomer especially, a short path from setup to first formulation matters more than an impressive feature list. As a concrete benchmark: a single formulation should take a couple of minutes, not an afternoon. If the routine workflow runs long, the system will quietly fall out of use.
There is a subtler trap here worth flagging: over-automation. Heavily automated platforms look reassuring in a demo, but the automation often hides the parameters you most need to control, adds failure points, and makes the system rigid and difficult to troubleshoot. Many otherwise capable systems become effectively unusable for exploratory research because the automation gets in the way rather than helping. This is also part of why IJM setups feel so heavy at the bench: the process machinery overwhelms the actual experiment. The sweet spot is control with simplicity, not control buried under it.
Typical newcomer pitfalls
A few patterns show up again and again with researchers buying their first formulation system. They are easy to avoid once named.
- Buying single-use without doing the yearly math. The per-run cost feels trivial until you multiply it by a year of weekly formulations.
- Choosing a screening-only system, then hitting a wall at in vivo. When the platform cannot reach the volumes your animal studies need, you end up re-optimizing everything on a second machine, and your carefully tuned screening particles do not carry over.
- Optimizing on a platform with no path forward. If there is no realistic route from your bench system toward larger scale, you may spend months perfecting a formulation that has to be rebuilt from scratch later.
- Trusting EE% alone. A high efficiency number can mask poor yield and heavy losses.
- Mistaking automation for capability. More automation is not more control, nor even sometimes higher throughput, due to the induced complexity.
The thread connecting all of these is the same idea from the start of this review: your nanoparticle is tied to your system, and transitions are where projects get expensive. Choosing with your development path in mind, not just today’s experiment, is what separates a good first decision from a costly one.
Where this leaves you
If you are early in the field and working at small volumes, microfluidic formulation is almost always the right starting point: it gives you control, reproducibility, and frugal use of RNA, and it spans the research range from screening through in vivo. The questions that then matter most are the practical ones above (real volume range, yield and losses, payload and lipid flexibility, reusable vs single-use, genuine reproducibility, and whether the system stays simple enough to actually run).
This is the logic behind TAMARA, Inside Therapeutics’ microfluidic formulation platform, which is built to cover the full R&D range (from a few hundred microliters of screening to in vivo-ready batches) on a single instrument with reusable chips and high RNA recovery. It is one example of a system designed around these parameters rather than around a single headline spec.
Whatever lipid nanoparticle formulation system you choose, come back to the idea we started with: your nanoparticle travels with your system. The particle is not a thing you own and move between machines; it is re-made every time the mixing changes, and in practice you will almost never get exactly the same nanoparticle back. No technology yet keeps it identical from screening all the way to production. That is precisely why the smart move is to choose for where your science is going, not only for the experiment in front of you: every avoidable transition is another particle you cannot fully reproduce, and another set of early results you may have to earn again from scratch.onizable lipids interact with and stabilize nucleic acids.
By adjusting the lipid types and ratios, researchers can modulate key nanoparticle properties such as particle size, surface charge, encapsulation efficiency, stability, biodistribution, and intracellular delivery performance. As a result, optimizing LNP composition has become a major challenge in the development of efficient nucleic acid therapeutics.

Looking to improve your LNP formulation using microfluidics?
Reach out to us to learn how we can help!
How do I formulate lipid nanoparticles?
RNA-LNPs are made by rapidly mixing an ethanolic lipid phase with an aqueous phase containing RNA. The rapid solvent exchange drives the lipids to self-assemble around the RNA into nanoparticles within milliseconds. The mixing method (hand mixing, microfluidics, or impingement jet mixing) determines how controlled and reproducible that assembly is, which is why the formulation system matters so much.
What is the difference between an LNP formulation system and an LNP formulation platform?
The terms are often used interchangeably. In practice, “system” or “platform” usually refers to the instrument and its associated mixing technology, chips or cartridges, and software used to produce nanoparticles in a controlled, repeatable way, as opposed to a manual bench method.
What is the best LNP formulation method for beginners?
For most newcomers, microfluidic formulation is the best starting point. It gives reproducible, controlled mixing on small volumes, which suits the low-input screening and in vitro work where people usually begin, and it uses little of your expensive RNA. Hand mixing is fine for a first taste of the field but quickly hits a reproducibility ceiling, while impingement jet mixing is built for large-scale production rather than the bench.
How much RNA do I need to formulate LNPs?
It depends on your stage. Physicochemical characterization and in vitro work typically need only a few hundred microliters per condition (around 200–500 µL), while mouse in vivo studies push you into the milliliter range (roughly 100 µL per mouse, times your cohort, plus QC). Larger in vivo studies can require tens of milliliters. The exact amount varies with your route of administration, working concentration, and indication.
Is microfluidics or impingement jet mixing better for RNA-LNP formulation?
Neither is universally better; they suit different stages. Microfluidics excels at control, reproducibility, and low-volume work, making it ideal for research and preclinical development. Impingement jet mixing excels at large-volume continuous production but is impractical at small scale. The right choice depends on the volumes you work with and where your project is heading.
What is the difference between encapsulation efficiency and encapsulation yield?
Encapsulation efficiency (EE%) is the fraction of RNA encapsulated in your final sample; a high EE% means little free RNA left over, which limits toxicity and reactogenicity. Encapsulation yield (EY%) is how much of the RNA you started with you actually recover encapsulated, accounting for losses during the run, so it behaves as a process parameter tied to cost and waste. A system can show high EE% while still wasting a large fraction of your input RNA, which is why both are worth checking.
Why do my nanoparticles change when I switch formulation systems?
Because particle formation is tightly coupled to mixing conditions. Different geometries and flow regimes produce different sizes, internal structures, and surface properties, even with identical lipids and ratios. This is why a formulation optimized on one system is not guaranteed to reproduce on another, and why no current technology keeps the same nanoparticle identical across every scale.
References
[1] T. Bethiana, A. Aljabbari, Y. Li, H. Mitra, M. Baghbanbashi, G. Harris, S.R. Dasaro, F. Masoomi, F. S. Vago, S. L. Hartzler, M. Figueiredo, L. A. Metskas, P. Vlachos, A. Ardekani, Y. Yeo, K. Ristroph bioRxiv 2025.11.07.687311; doi: https://doi.org/10.1101/2025.11.07.687311



